Publications
My articles and reviews
2024
- Chem Catalysis
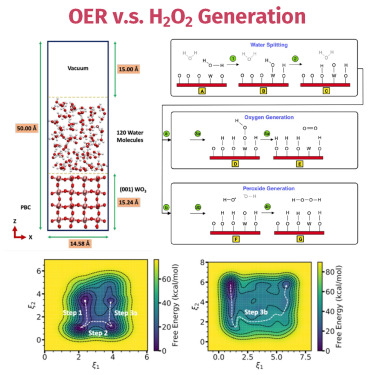 A Metadynamics Study of Water Oxidation Reactions at (001)-WO₃/Liquid-Water InterfaceRangsiman Ketkaew, Fabrizio Creazzo, Kevin Sivula, and 1 more authorChem Catalysis, Sep 2024
A Metadynamics Study of Water Oxidation Reactions at (001)-WO₃/Liquid-Water InterfaceRangsiman Ketkaew, Fabrizio Creazzo, Kevin Sivula, and 1 more authorChem Catalysis, Sep 2024@article{ketkaew_metadynamics_2024, title = {A Metadynamics Study of Water Oxidation Reactions at (001)-{WO₃}/Liquid-Water Interface}, volume = {4}, issn = {2667-1107, 2667-1093}, url = {https://www.cell.com/chem-catalysis/abstract/S2667-1093(24)00257-4}, doi = {10.1016/j.checat.2024.101085}, language = {English}, number = {9}, urldate = {2024-10-11}, journal = {Chem Catalysis}, author = {Ketkaew, Rangsiman and Creazzo, Fabrizio and Sivula, Kevin and Luber, Sandra}, month = sep, year = {2024}, keywords = {and infrastructure, collective variables, innovation, machine learning, metadynamics, neural network, SDG9: Industry, tungsten trioxide, water oxidation reactions}, } - Chem. Eur. J.
 Machine Learning Interatomic Potentials for CatalysisDeqi Tang , Rangsiman Ketkaew, and Sandra LuberChemistry - A European Journal, Sep 2024
Machine Learning Interatomic Potentials for CatalysisDeqi Tang , Rangsiman Ketkaew, and Sandra LuberChemistry - A European Journal, Sep 2024Atomistic modeling can provide insights into the design of novel catalysts in modern industries of chemistry, materials science, and biology. Classical force fields and ab initio calculations have been widely adopted in molecular simulations. How- ever, these methods suffer from the drawbacks of either low accuracy or high cost. Recently, the development of machine learning interatomic potentials (MLIPs) has become more and more popular as they can tackle the problems in question and can deliver rather accurate results at significantly lower computational cost. In this review, the atomistic modeling of catalytic systems with the aid of MLIPs is discussed, showcasing recently developed MLIP models and selected applications for the modeling of catalytic systems. We also highlight the best practices, and challenges for MLIPs and give an outlook for future works on MLIPs in the field of catalysis.
@article{tang_machine_nodate, title = {Machine {Learning} {Interatomic} {Potentials} for {Catalysis}}, copyright = {© 2024 Wiley-VCH GmbH}, issn = {1521-3765}, url = {https://onlinelibrary.wiley.com/doi/abs/10.1002/chem.202401148}, doi = {10.1002/chem.202401148}, language = {en}, urldate = {2024-10-11}, journal = {Chemistry - A European Journal}, author = {Tang, Deqi and Ketkaew, Rangsiman and Luber, Sandra}, month = sep, year = {2024}, keywords = {Computational chemistry, Heterogeneous Catalysis, Homogeneous catalysis, MLIPs, molecular dynamics}, pages = {e202401148}, category = {review}, }
2023
- Chimia
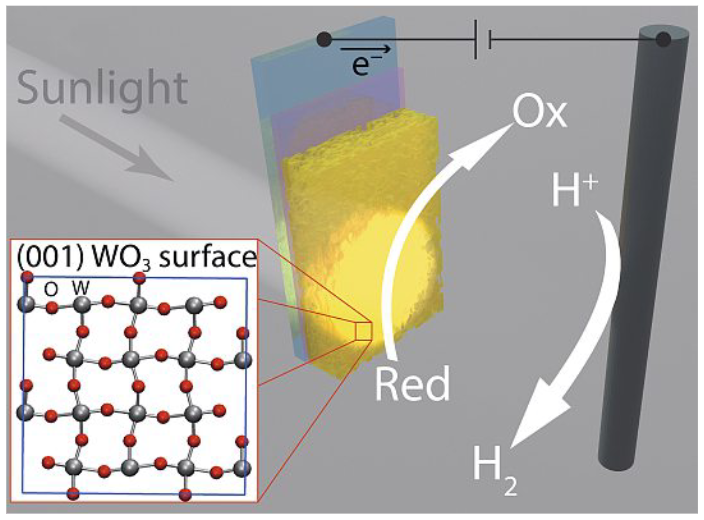 Enabling Direct Photoelectrochemical H₂ Production using Alternative Oxidation Reactions on WO₃Nukorn Plainpan , Rangsiman Ketkaew, Sandra Luber, and 1 more authorChimia, Mar 2023
Enabling Direct Photoelectrochemical H₂ Production using Alternative Oxidation Reactions on WO₃Nukorn Plainpan , Rangsiman Ketkaew, Sandra Luber, and 1 more authorChimia, Mar 2023The efficient and inexpensive conversion of solar energy into chemical bonds, such as in H2 via the photoelectrochemical splitting of H2O, is a promising route to produce green industrial feedstocks and renewable fuels, which is a key goal of the NCCR Catalysis. However, the oxidation product of the water splitting reaction, O2, has little economic or industrial value. Thus, upgrading key chemical species using alternative oxidation reactions is an emerging trend. WO₃ has been identified as a unique photoanode material for this purpose since it performs poorly in the oxygen evolution reaction in H2O. Herein we highlight a collaboration in the NCCR Catalysis that has gained insights at the atomic level of the WO₃ surface with ab initio computational methods that help to explain its unique catalytic activity. These computational efforts give new context to experimental results employing WO₃ photoanodes for the direct photoelectrochemical oxidation of biomass-derived 5-(hydroxymethyl) furfural. While yield for the desired product, 2,5-furandicarboxylic acid is low, insights into the reaction rate constants using kinetic modelling and an electrochemical technique called derivative voltammetry, give indications on how to improve the system.
@article{plainpan_enabling_2023, title = {Enabling {Direct} {Photoelectrochemical} {H}₂ {Production} using {Alternative} {Oxidation} {Reactions} on {WO}₃}, volume = {77}, copyright = {info:eu-repo/semantics/openAccess}, issn = {0009-4293}, url = {https://www.chimia.ch/chimia/article/view/2023_110}, doi = {10.2533/chimia.2023.110}, language = {eng}, number = {3}, urldate = {2024-10-11}, journal = {Chimia}, author = {Plainpan, Nukorn and Ketkaew, Rangsiman and Luber, Sandra and Sivula, Kevin}, month = mar, year = {2023}, pmid = {38047812}, pages = {110--115}, category = {review}, }
2022
- JCIM
 DeepCV: A Deep Learning Framework for Blind Search of Collective Variables in Expanded Configurational SpaceRangsiman Ketkaew, and Sandra LuberJ. Chem. Inf. Model., Dec 2022
DeepCV: A Deep Learning Framework for Blind Search of Collective Variables in Expanded Configurational SpaceRangsiman Ketkaew, and Sandra LuberJ. Chem. Inf. Model., Dec 2022We present Deep learning for Collective Variables (DeepCV), a computer code that provides an efficient and customizable implementation of the deep autoencoder neural network (DAENN) algorithm that has been developed in our group for computing collective variables (CVs) and can be used with enhanced sampling methods to reconstruct free energy surfaces of chemical reactions. DeepCV can be used to conveniently calculate molecular features, train models, generate CVs, validate rare events from sampling, and analyze a trajectory for chemical reactions of interest. We use DeepCV in an example study of the conformational transition of cyclohexene, where metadynamics simulations are performed using DAENN-generated CVs. The results show that the adopted CVs give free energies in line with those obtained by previously developed CVs and experimental results. DeepCV is open-source software written in Python/C++ object-oriented languages, based on the TensorFlow framework and distributed free of charge for noncommercial purposes, which can be incorporated into general molecular dynamics software. DeepCV also comes with several additional tools, i.e., an application program interface (API), documentation, and tutorials.
@article{ketkaew_deepcv_2022, title = {{DeepCV}: {A} {Deep} {Learning} {Framework} for {Blind} {Search} of {Collective} {Variables} in {Expanded} {Configurational} {Space}}, volume = {62}, issn = {1549-9596}, shorttitle = {{DeepCV}}, url = {https://doi.org/10.1021/acs.jcim.2c00883}, doi = {10.1021/acs.jcim.2c00883}, number = {24}, urldate = {2024-10-11}, journal = {J. Chem. Inf. Model.}, author = {Ketkaew, Rangsiman and Luber, Sandra}, month = dec, year = {2022}, pages = {6352--6364}, } - Appl. Surf. Sci.
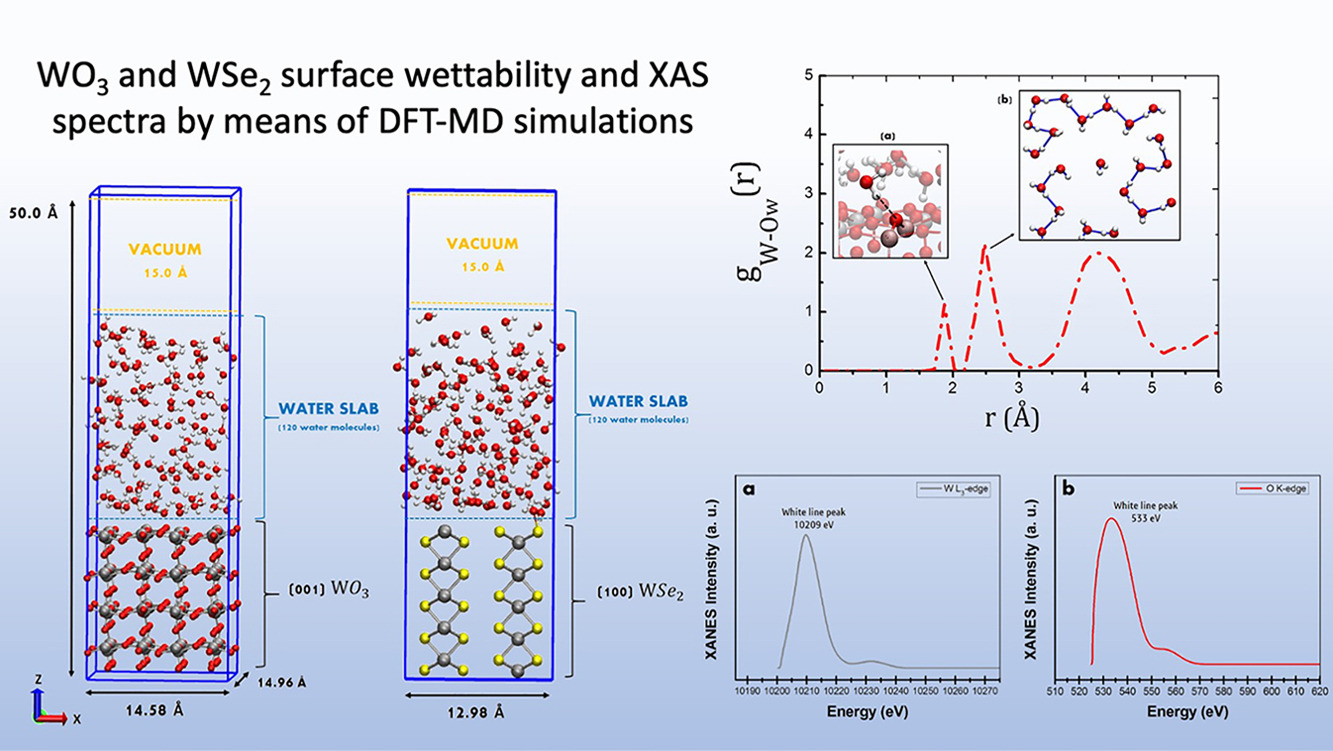 Effects of surface wettability on (001)-WO₃ and (100)-WSe₂: A spin-polarized DFT-MD studyFabrizio Creazzo , Rangsiman Ketkaew, Kevin Sivula, and 1 more authorApplied Surface Science, Nov 2022
Effects of surface wettability on (001)-WO₃ and (100)-WSe₂: A spin-polarized DFT-MD studyFabrizio Creazzo , Rangsiman Ketkaew, Kevin Sivula, and 1 more authorApplied Surface Science, Nov 2022An extensive understanding of WO₃ and WSe₂ bulk crystalline structures and explicit solvent effects on (001)-WO₃ and (100)-WSe₂ facets are essential for design of efficient (photo) electrocatalysts. The atomistic level understanding of both WO₃ and WSe₂ bulk solids and how water solvation processes occur on WO₃ and WSe₂ facets are nowadays characterized by a noticeable lack of knowledge. Herein, forefront Density Functional Theory-based molecular dynamics have been conducted for assessing the role of an explicit water environment in the characterization of solid surfaces. Water at the interface and H-bonds environment, as well as WO₃ and WSe₂ surface activity, will be described in terms of surface wettability and interfacial water dynamics, revealing the relevance of treating explicitly liquid water and its dynamics in assessing catalytic features. We provide pieces of evidence of the hydrophobic character shown by (001)-WO₃ and (100)-WSe₂ facets. A preferential in-plane hydration structure of the first water layer has been detected at both (001)-WO₃ and (100)-WSe₂ water interface, in which the electric dipole moment of water molecules is re-oriented in a sort of 2-dimensional H-bond network. Bulk property calculations of WO₃ and WSe₂ are also provided.
@article{creazzo_effects_2022, title = {Effects of surface wettability on (001)-WO₃ and (100)-WSe₂: A spin-polarized DFT-MD study}, volume = {601}, issn = {0169-4332}, url = {https://www.sciencedirect.com/science/article/pii/S016943322201738X}, doi = {10.1016/j.apsusc.2022.154203}, urldate = {2024-10-11}, journal = {Applied Surface Science}, author = {Creazzo, Fabrizio and Ketkaew, Rangsiman and Sivula, Kevin and Luber, Sandra}, month = nov, year = {2022}, keywords = {DFT-MD, Tungsten oxide WO, Tungsten selenide WSe, Water, X-ray absorption spectroscopy}, pages = {154203}, } - ChemSusChem
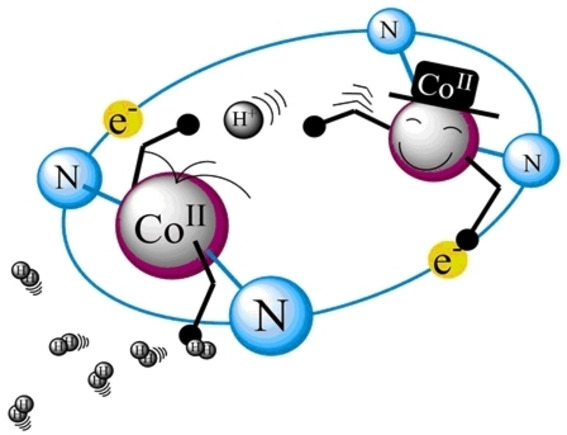 Two Novel Dinuclear Cobalt Polypyridyl Complexes in Electro- and Photocatalysis for Hydrogen Production: Cooperativity Increases PerformanceNicola Weder, Nora S. Grundmann, Benjamin Probst, and 5 more authorsChemSusChem, Nov 2022
Two Novel Dinuclear Cobalt Polypyridyl Complexes in Electro- and Photocatalysis for Hydrogen Production: Cooperativity Increases PerformanceNicola Weder, Nora S. Grundmann, Benjamin Probst, and 5 more authorsChemSusChem, Nov 2022Syntheses and mechanisms of two dinuclear Co-polypyridyl catalysts for the H2 evolution reaction (HER) were reported and compared to their mononuclear analogue (R1). In both catalysts, two di-(2,2’-bipyridin-6-yl)-methanone units were linked by either 2,2’-bipyridin-6,6’-yl or pyrazin-2,5-yl. Complexation with CoII gave dinuclear compounds bridged by pyrazine (C2) or bipyridine (C1). Photocatalytic HER gave turnover numbers (TONs) of up to 20000 (C2) and 7000 (C1) in water. Electrochemically, C1 was similar to the R1, whereas C2 showed electronic coupling between the two Co centers. The E(CoII/I) split by 360 mV into two separate waves. Proton reduction in DMF was investigated for R1 with [HNEt3](BF4) by simulation, foot of the wave analysis, and linear sweep voltammetry (LSV) with in-line detection of H2. All methods agreed well with an (E)ECEC mechanism and the first protonation being rate limiting (≈104 m−1 s−1). The second reduction was more anodic than the first one. pKa values of around 10 and 7.5 were found for the two protonations. LSV analysis with H2 detection for all catalysts and acids with different pKa values [HBF4, pKa(DMF)≈3.4], intermediate [HNEt3](BF4), pKa(DMF)≈9.2 to weak [AcOH, pKa(DMF)≈13.5] confirmed electrochemical H2 production, distinctly dependent on the pKa values. Only HBF4 protonated CoI intermediates. The two metals in the dualcore C2 cooperated with an increase in rate to a competitive 105 m−1 s−1 with [HNEt3](BF4). The overpotential decreased compared to R1 by 100 mV. Chronoamperometry established high stabilities for all catalysts with TONlim of 100 for R1 and 320 for C1 and C2.
@article{weder_two_2022, title = {Two {Novel} {Dinuclear} {Cobalt} {Polypyridyl} {Complexes} in {Electro}- and {Photocatalysis} for {Hydrogen} {Production}: {Cooperativity} {Increases} {Performance}}, volume = {15}, copyright = {© 2022 The Authors. ChemSusChem published by Wiley-VCH GmbH}, issn = {1864-564X}, url = {https://onlinelibrary.wiley.com/doi/abs/10.1002/cssc.202201049}, doi = {10.1002/cssc.202201049}, language = {en}, number = {17}, urldate = {2024-10-11}, journal = {ChemSusChem}, author = {Weder, Nicola and Grundmann, Nora S. and Probst, Benjamin and Blacque, Olivier and Ketkaew, Rangsiman and Creazzo, Fabrizio and Luber, Sandra and Alberto, Roger}, year = {2022}, keywords = {artificial photosynthesis, electrocatalysis, hydrogen, solar fuels, water reduction}, pages = {e202201049}, } - ChemRxiv
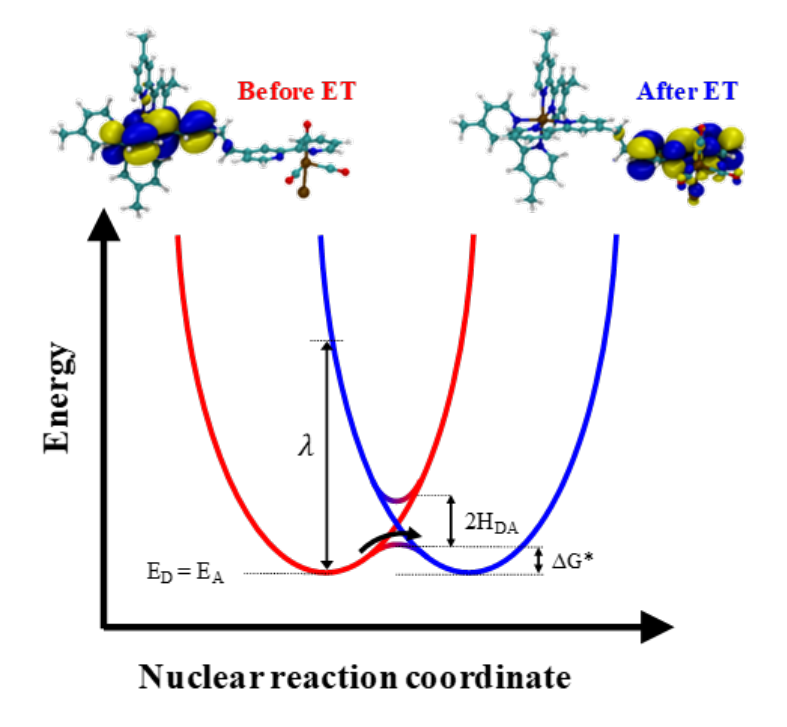 Electronic Communication and Photoinduced Intramolecular Electron Transfer in Hybrid Ru(II)-Re(I) Complexes Using Eigenstate-Based and Diabatic-State-Based ModelsRangsiman KetkaewMar 2022
Electronic Communication and Photoinduced Intramolecular Electron Transfer in Hybrid Ru(II)-Re(I) Complexes Using Eigenstate-Based and Diabatic-State-Based ModelsRangsiman KetkaewMar 2022Photoinduced intramolecular electron transfer (PIET) plays a vital role in the efficiency of electronics communication in transition metal complexes catalysing oxidation-reduction reaction. In this work, we theoretically calculate the rate of electron transfer(ET) in Ru(II)-BL-Ru(I) hybrid complexes; where BL is bridging ligand. A brief concept of ET in the basis of Marcus theory, which is extended to address a variety of different type of ET, is provided. We show that, in the case of Ru(II)-BL-Ru(I) complex, ET involves a non-adiabatic state which thanks to a fast electronics communication between donor and acceptor connected by BL and becomes rigid complex. Single electron transferring in Ru(II)-BL-Ru(I) complex governed by PIET constructed by potential energy curve as change of structural transformation over time-evolution. We also investigate the mechanism of PIET involving a redox reaction in excited state, wherein the oxidation state of Ru(II) (donor) and Ru(I) (acceptor) changes. To access non-adiabatic state of Ru(II)-BL-Ru(I), we use constrained density functional theory to allow ground state calculation to be performed along with geometry constraints. We also systematically study the role of distance of donor-acceptor separation on kinetics of PIET
@misc{ketkaew_electronic_2022, title = {Electronic {Communication} and {Photoinduced} {Intramolecular} {Electron} {Transfer} in {Hybrid} {Ru}({II})-{Re}({I}) {Complexes} {Using} {Eigenstate}-{Based} and {Diabatic}-{State}-{Based} {Models}}, url = {https://chemrxiv.org/engage/chemrxiv/article-details/623edf74ab00517a8099e65f}, doi = {10.26434/chemrxiv-2022-40c31-v3}, journal = {ChemRxiv}, language = {en}, urldate = {2024-10-11}, publisher = {ChemRxiv}, author = {Ketkaew, Rangsiman}, month = mar, year = {2022}, keywords = {Constrained Density Functional Theory, Intramolecular electron transfer, Marcus theory rate constants, Photoinduced electron transfer}, } - JPCL
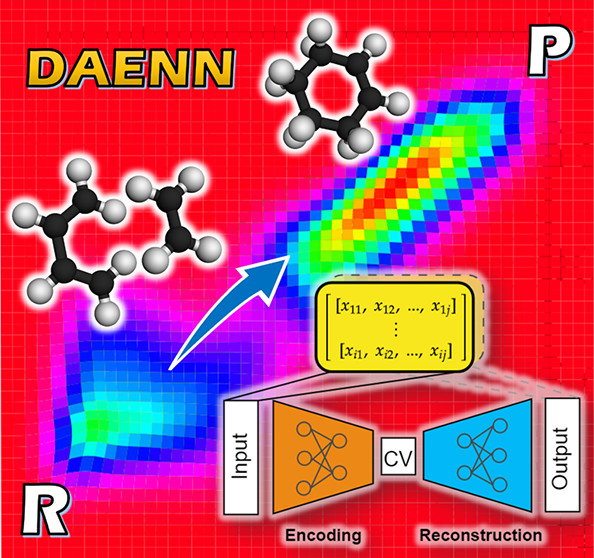 Machine Learning-Assisted Discovery of Hidden States in Expanded Free Energy SpaceRangsiman Ketkaew, Fabrizio Creazzo, and Sandra LuberJ. Phys. Chem. Lett., Feb 2022
Machine Learning-Assisted Discovery of Hidden States in Expanded Free Energy SpaceRangsiman Ketkaew, Fabrizio Creazzo, and Sandra LuberJ. Phys. Chem. Lett., Feb 2022Collective variables (CVs) are crucial parameters in enhanced sampling calculations and strongly impact the quality of the obtained free energy surface. However, many existing CVs are unique to and dependent on the system they are constructed with, making the developed CV non-transferable to other systems. Herein, we develop a non-instructor-led deep autoencoder neural network (DAENN) for discovering general-purpose CVs. The DAENN is used to train a model by learning molecular representations upon unbiased trajectories that contain only the reactant conformers. The prior knowledge of nonconstraint reactants coupled with the here-introduced topology variable and loss-like penalty function are only required to make the biasing method able to expand its configurational (phase) space to unexplored energy basins. Our developed autoencoder is efficient and relatively inexpensive to use in terms of a priori knowledge, enabling one to automatically search for hidden CVs of the reaction of interest.
@article{ketkaew_machine_2022, title = {Machine {Learning}-{Assisted} {Discovery} of {Hidden} {States} in {Expanded} {Free} {Energy} {Space}}, volume = {13}, url = {https://doi.org/10.1021/acs.jpclett.1c04004}, doi = {10.1021/acs.jpclett.1c04004}, number = {7}, urldate = {2022-02-24}, journal = {J. Phys. Chem. Lett.}, author = {Ketkaew, Rangsiman and Creazzo, Fabrizio and Luber, Sandra}, month = feb, year = {2022}, pages = {1797--1805}, } - JPCA
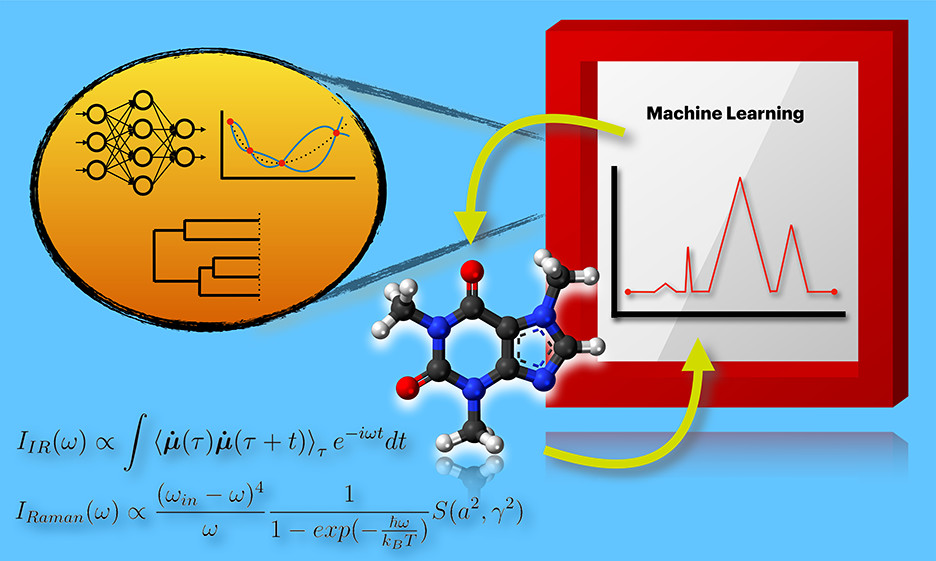 A Concise Review on Recent Developments of Machine Learning for the Prediction of Vibrational SpectraRuocheng Han* , Rangsiman Ketkaew*, and Sandra LuberJ. Phys. Chem. A, Feb 2022Selected as a journal cover
A Concise Review on Recent Developments of Machine Learning for the Prediction of Vibrational SpectraRuocheng Han* , Rangsiman Ketkaew*, and Sandra LuberJ. Phys. Chem. A, Feb 2022Selected as a journal coverMachine learning has become more and more popular in computational chemistry, as well as in the important field of spectroscopy. In this concise review, we walk the reader through a short summary of machine learning algorithms and a comprehensive discussion on the connection between machine learning methods and vibrational spectroscopy, particularly for the case of infrared and Raman spectroscopy. We also briefly discuss state-of-the-art molecular representations which serve as meaningful inputs for machine learning to predict vibrational spectra. In addition, this review provides an overview of the transferability and best practices of machine learning in the prediction of vibrational spectra as well as possible future research directions.
@article{han_concise_2022, title = {A {Concise} {Review} on {Recent} {Developments} of {Machine} {Learning} for the {Prediction} of {Vibrational} {Spectra}}, volume = {126}, issn = {1089-5639}, url = {https://doi.org/10.1021/acs.jpca.1c10417}, doi = {10.1021/acs.jpca.1c10417}, number = {6}, urldate = {2024-10-11}, journal = {J. Phys. Chem. A}, author = {Han, Ruocheng and Ketkaew, Rangsiman and Luber, Sandra}, month = feb, year = {2022}, note = {Selected as a journal cover}, pages = {801--812}, category = {review}, } - Top. Catal.
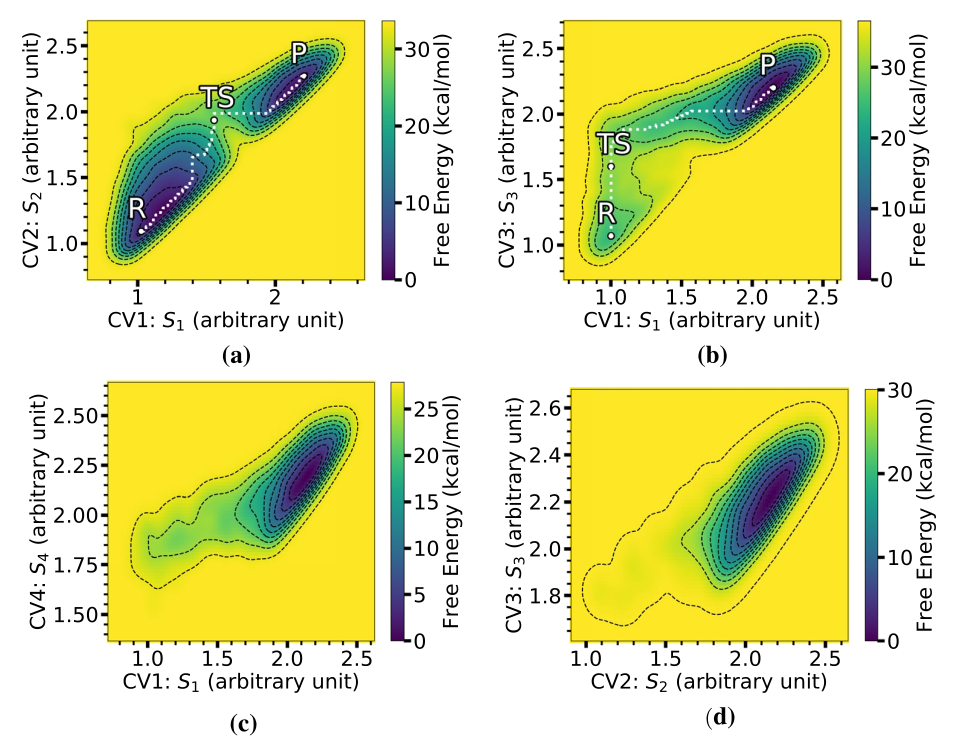 Closer Look at Inverse Electron Demand Diels–Alder and Nucleophilic Addition Reactions on s-Tetrazines Using Enhanced Sampling MethodsRangsiman Ketkaew, Fabrizio Creazzo, and Sandra LuberTopics in Catalysis, Feb 2022
Closer Look at Inverse Electron Demand Diels–Alder and Nucleophilic Addition Reactions on s-Tetrazines Using Enhanced Sampling MethodsRangsiman Ketkaew, Fabrizio Creazzo, and Sandra LuberTopics in Catalysis, Feb 2022Inverse electron demand [4+2] Diels–Alder (iEDDA) reactions as well as unprecedented nucleophilic (azaphilic) additions of R-substituted silyl-enol ethers (where R is Phenyl, Methyl, and Hydrogen) to 1,2,4,5-tetrazine (s-tetrazine) catalyzed by \{}hbox {BF}_{3}\\have recently been discovered (Simon et al. in Org Lett 23(7):2426–2430, 2021), where static calculations were employed for calculation of activation energies. In order to have a more realistic dynamic description of these reactions in explicit solution at ambient conditions, in this work we use a semiempirical tight-binding method combined with enhanced sampling techniques to calculate free energy surfaces (FESs) of the iEDDA and azaphilic addition reactions. Relevant products of not only s-tetrazine but also its derivatives such as \{}hbox {BF}_{3}\\-mediated s-tetrazine adducts are investigated. We reconstruct the FESs of the iEDDA and azaphilic addition reactions using metadynamics and blue moon ensemble, and compare the ability of different collective variables (CVs) including bond distances, Social PeRmutation INvarianT (SPRINT) coordinates, and path-CV to describe the reaction pathway. We find that when a bulky Phenyl is used as a substituent at the dienophile the azaphilic addition is preferred over the iEDDA reaction. In addition, we also investigate the effect of \{}hbox {BF}_{3}\∈the diene and steric hindrance in the dienophile on the competition between the iEDDA and azaphilic addition reactions, providing chemical insight for reaction design.
@article{ketkaew_closer_2022, title = {Closer {Look} at {Inverse} {Electron} {Demand} {Diels}–{Alder} and {Nucleophilic} {Addition} {Reactions} on s-{Tetrazines} {Using} {Enhanced} {Sampling} {Methods}}, volume = {65}, issn = {1572-9028}, url = {https://doi.org/10.1007/s11244-021-01516-y}, doi = {10.1007/s11244-021-01516-y}, language = {en}, number = {1}, urldate = {2024-10-11}, journal = {Topics in Catalysis}, author = {Ketkaew, Rangsiman and Creazzo, Fabrizio and Luber, Sandra}, month = feb, year = {2022}, keywords = {Azaphilic addition, Blue moon ensemble, iEDDA reaction, Metadynamics}, pages = {1--17}, }
2021
- Chimia
 How ab initio Molecular Dynamics Can Change the Understanding on Transition Metal Catalysed Water OxidationMauro Schilling* , Rangsiman Ketkaew*, and Sandra LuberCHIMIA, Mar 2021
How ab initio Molecular Dynamics Can Change the Understanding on Transition Metal Catalysed Water OxidationMauro Schilling* , Rangsiman Ketkaew*, and Sandra LuberCHIMIA, Mar 2021Artificial water splitting is a promising technology that allows the storage of renewable energy in the form of energy-rich compounds. This mini-review showcases how theoretical studies contribute to the under-standing of existing water oxidation catalysts (WOCs) as well as inspiring the development of novel WOCs. In order to understand the chemical complexity of transition metal complexes and their interaction with the solvent environment, the use of sophisticated simulation protocols is necessary. As an illustration, a family of ruthe- nium-based WOCs is presented which were investigated employing a wide range of forefront computational methods with emphasis on ab initiomolecular dynamic based approaches. In those studies a base assisted oxygen–oxygen bond formation was identified as the energetically most favourable reaction mechanism. By examining the role of local environmental effects at ambient temperature and the effect of modifications in the ligand framework, a comprehensible picture of the WOCs can be given, where the latter can serve as a guideline for further experimental and computational studies. In this mini-review, we provide a description of the methods, and the findings of our previous computational studies in compacted form, aimed at scientists with a theoretical as well as experimental background.
@article{schilling_how_2021, title = {How ab initio {Molecular} {Dynamics} {Can} {Change} the {Understanding} on {Transition} {Metal} {Catalysed} {Water} {Oxidation}}, volume = {75}, copyright = {Copyright (c) 2021 Mauro Schilling, Rangsiman Ketkaew, Sandra Luber}, issn = {2673-2424}, url = {https://www.chimia.ch/chimia/article/view/2021_195}, doi = {10.2533/chimia.2021.195}, language = {en}, number = {3}, urldate = {2024-10-11}, journal = {CHIMIA}, author = {Schilling, Mauro and Ketkaew, Rangsiman and Luber, Sandra}, month = mar, year = {2021}, keywords = {Density functional theory, Enhanced sampling, Molecular dynamics, Transition metal catalysis, Water oxidation}, pages = {195--195}, category = {review}, } - Dalton Trans.
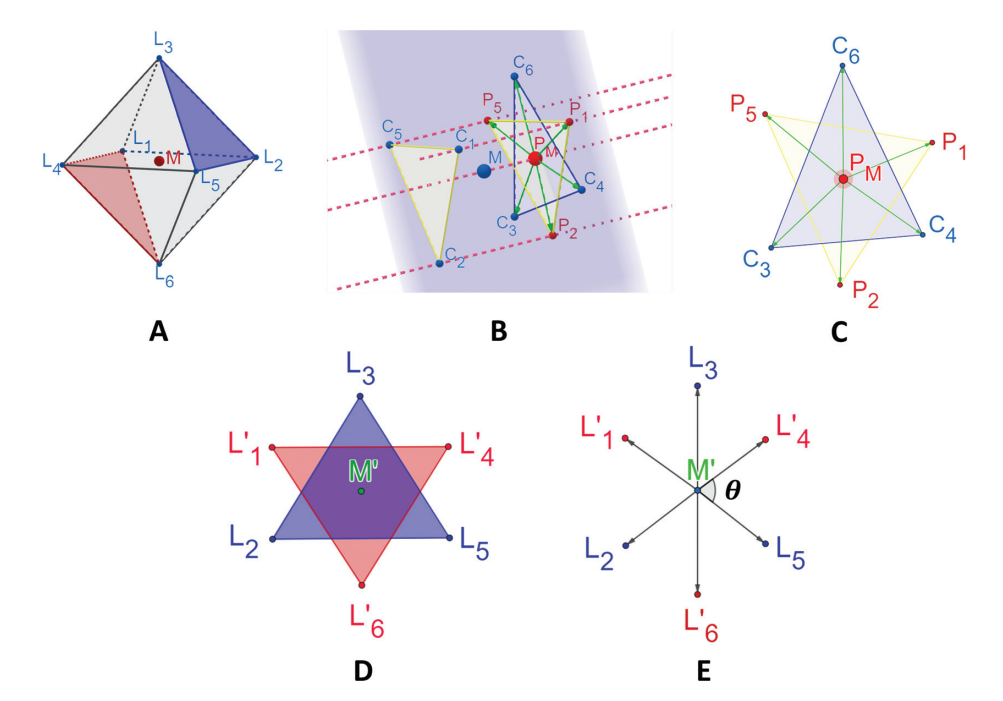 OctaDist: A Tool for Calculating Distortion Parameters in Spin Crossover and Coordination ComplexesRangsiman Ketkaew, Yuthana Tantirungrotechai, Phimphaka Harding, and 4 more authorsDalton Transactions. More Information can be found here , Mar 2021
OctaDist: A Tool for Calculating Distortion Parameters in Spin Crossover and Coordination ComplexesRangsiman Ketkaew, Yuthana Tantirungrotechai, Phimphaka Harding, and 4 more authorsDalton Transactions. More Information can be found here , Mar 2021OctaDist is an interactive and visual program for determination of structural distortion in octahedral coordination complexes such as spin crossover complexes, single-ion magnets, perovskites or metal–organic frameworks. OctaDist computes the octahedral distortion parameters initially designed in the context of the spin-crossover phenomenon and denoted ζ, Σ, and Θ from standard structural files. The program also provides additional tools for molecular analyses and visualization. It emphasizes performance, flexibility, ease of use, application programming interface (API) consistency, and clear documentation. The modules and classes in OctaDist can be easily customized to include new algorithms or analytical tools. OctaDist is cross-platform supported for modern operating systems and is available as open-source distributed under the GNU General Public License version 3.
@article{ketkaew_octadist_2021, title = {{OctaDist}: A Tool for Calculating Distortion Parameters in Spin Crossover and Coordination Complexes}, volume = {50}, shorttitle = {{OctaDist}}, url = {https://pubs.rsc.org/en/content/articlelanding/2021/dt/d0dt03988h}, doi = {10.1039/D0DT03988H}, language = {en}, number = {3}, urldate = {2024-10-11}, journal = {Dalton Transactions}, author = {Ketkaew, Rangsiman and Tantirungrotechai, Yuthana and Harding, Phimphaka and Chastanet, Guillaume and Guionneau, Philippe and Marchivie, Mathieu and J. Harding, David}, year = {2021}, pages = {1086--1096}, }
2018
- Macromol. Theory Simul.
 Dissipative Particle Dynamics Study of SWCNT Reinforced Natural Rubber Composite System: An Important Role of Self-Avoiding Model on Mechanical PropertiesRangsiman Ketkaew, and Yuthana TantirungrotechaiMacromolecular Theory and Simulations, Mar 2018
Dissipative Particle Dynamics Study of SWCNT Reinforced Natural Rubber Composite System: An Important Role of Self-Avoiding Model on Mechanical PropertiesRangsiman Ketkaew, and Yuthana TantirungrotechaiMacromolecular Theory and Simulations, Mar 2018Dissipative particle dynamics (DPD) is used to predict the mechanical properties of crosslinking-polyisoprene (PI) reinforced by single-walled carbon nanotube (SWCNT). The role of the increasing concentration of SWCNT up to 8% in morphology of PI composite is investigated. The carbon nanotube (CNT) restricts the polymer movement as indicated by the reduction of mean square displacement and the end-to-end distance. The analysis of CNT bundle reveals that the CNT forms bundle of many sizes depending on its concentration. The mechanical reinforcement of the PI composites is attributed to the restricted motion of crosslinked entangled polyisoprene and to the alignment of aggregated CNT along the elongation direction. However, the DPD alone fails to account for the change in Young’s modulus of the composite system as a function of the SWCNT concentration. Using the modified segmental repulsive potential (mSRP), which prevents polymeric chain crossing and enhances polymer entanglement, can address this shortcoming. The mSRP is necessary for correct description of mechanical reinforcement in a polymer composite system. To obtain quantitative agreement with the experimental modulus, the polyisoprene interaction parameter is modified from the published value by 10%. This modified parameter is found to work well for the simulation of polyisoprene composites.
@article{ketkaew_dissipative_2018, title = {Dissipative {Particle} {Dynamics} {Study} of {SWCNT} {Reinforced} {Natural} {Rubber} {Composite} {System}: {An} {Important} {Role} of {Self}-{Avoiding} {Model} on {Mechanical} {Properties}}, volume = {27}, issn = {1521-3919}, shorttitle = {Dissipative {Particle} {Dynamics} {Study} of {SWCNT} {Reinforced} {Natural} {Rubber} {Composite} {System}}, url = {https://onlinelibrary.wiley.com/doi/abs/10.1002/mats.201700093}, doi = {10.1002/mats.201700093}, language = {en}, number = {3}, urldate = {2024-10-11}, journal = {Macromolecular Theory and Simulations}, author = {Ketkaew, Rangsiman and Tantirungrotechai, Yuthana}, year = {2018}, keywords = {dissipative particle dynamics, mechanical properties, molecular dynamics, nanocomposites, rubber}, pages = {1700093}, } - Dalton Trans.
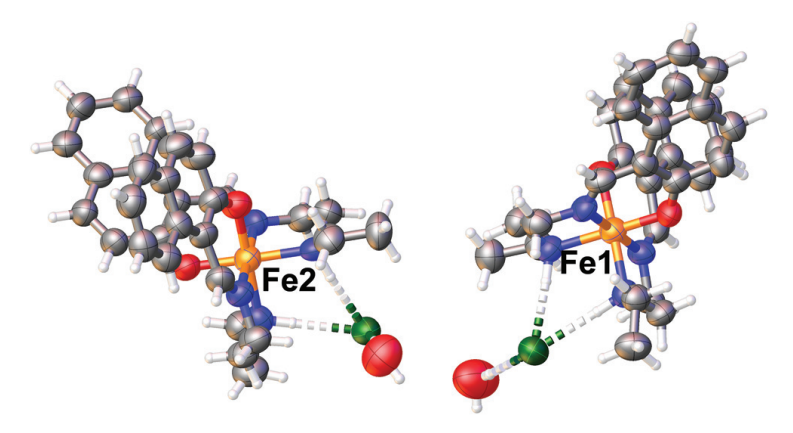 Solvatomorphism and Anion Effects in Predominantly Low Spin Iron(iii) Schiff Base ComplexesTheerapoom Boonprab, Phimphaka Harding, Keith S. Murray, and 7 more authorsDalton Transactions, Mar 2018
Solvatomorphism and Anion Effects in Predominantly Low Spin Iron(iii) Schiff Base ComplexesTheerapoom Boonprab, Phimphaka Harding, Keith S. Murray, and 7 more authorsDalton Transactions, Mar 2018A series of iron(III) complexes [Fe(naphEen)2]X·sol (naphEen = 1-[2-(ethylamino)-ethylimino]methyl-2-naphtholate; X = F, sol = 0.5CH2Cl2·H2O 1; sol = H2O, X = Cl, 2 and X = Br 3) and [Fe(naphEen)2]I 4 has been prepared. The UV-Vis spectra reveal clear differences for 1 which DFT/TDDFT calculations suggest are due to an equilibrium between [Fe(naphEen)2]F and [Fe(naphEen)2F], the latter having a coordinated F ligand. The X-ray crystal structures of 2–4 show LS Fe(III) centres in all cases and extensive aryl interactions that link the Fe centres into supramolecular squares. In 3 at room temperature the compound loses half an equivalent of water resulting in a change in space group from Monoclinic P21/n to C2/c. Magnetic studies indicate that 1 is trapped in a mixed spin state being ca. 40% HS while 2–4 are effectively low spin up to 350 K. In contrast, Mössbauer spectroscopic studies of 1 indicate a gradual but incomplete spin crossover. The magnetic properties of 2–4 contrast with the related [Fe(salEen-X)2]anion derivatives which are often spin crossover active.
@article{boonprab_solvatomorphism_2018, title = {Solvatomorphism and Anion Effects in Predominantly Low Spin Iron(iii) {Schiff} Base Complexes}, volume = {47}, url = {https://pubs.rsc.org/en/content/articlelanding/2018/dt/c8dt02016g}, doi = {10.1039/C8DT02016G}, language = {en}, number = {35}, urldate = {2024-10-11}, journal = {Dalton Transactions}, author = {Boonprab, Theerapoom and Harding, Phimphaka and S. Murray, Keith and Phonsri, Wasinee and G. Telfer, Shane and Alkaş, Adil and Ketkaew, Rangsiman and Tantirungrotechai, Yuthana and L. Jameson, Guy N. and J. Harding, David}, year = {2018}, pages = {12449--12458}, }
2017
- Catal. Sci. Technol.
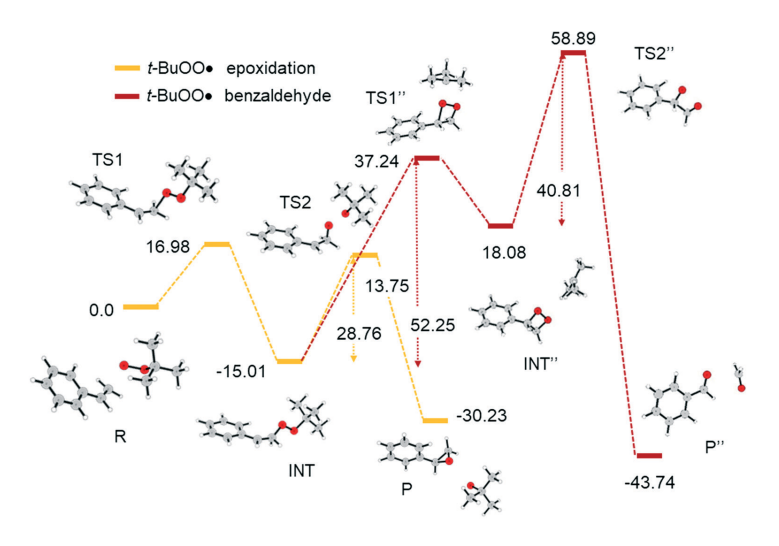 Microwave-Assisted One-Pot Functionalization of Metal-Organic Framework MIL-53(Al)-NH 2 with Copper(ii) Complexes and Its Application in Olefin OxidationT. Bunchuay, R. Ketkaew, P. Chotmongkolsap, and 4 more authorsCatalysis Science & Technology, Mar 2017
Microwave-Assisted One-Pot Functionalization of Metal-Organic Framework MIL-53(Al)-NH 2 with Copper(ii) Complexes and Its Application in Olefin OxidationT. Bunchuay, R. Ketkaew, P. Chotmongkolsap, and 4 more authorsCatalysis Science & Technology, Mar 2017A simple, rapid, and efficient functionalization of the metal-organic framework MIL-53(Al)-NH2 with copper(II) complexes by microwave irradiation is described. The one-pot post-synthetic modification was based on the Schiff base reaction between the aldehyde added and the primary aryl amino group in the organic linker of MIL-53(Al)-NH2, which afforded coordination sites for copper(II). The effects of synthetic parameters such as solvent, reaction time, and reactant ratio were evaluated. The functionalized materials were structurally characterized and examined for potential applications in catalysis. The copper complex functionalized MIL-53(Al) was active towards the oxidation of olefins with tert-butyl hydroperoxide as an oxidant. The results showed that if the olefin possessed allylic hydrogens such as cyclohexene, allylic oxidation products were obtained, but epoxide was the major product obtained from terminal olefins without allylic hydrogens. The reaction mechanism based on density functional theory calculations provides some insights into the observed experimental results.
@article{bunchuay_microwave-assisted_2017, title = {Microwave-Assisted One-Pot Functionalization of Metal-Organic Framework {MIL}-53({Al})-{NH} 2 with Copper(ii) Complexes and Its Application in Olefin Oxidation}, volume = {7}, url = {https://pubs.rsc.org/en/content/articlelanding/2017/cy/c7cy01941f}, doi = {10.1039/C7CY01941F}, language = {en}, number = {24}, urldate = {2024-10-11}, journal = {Catalysis Science \& Technology}, author = {Bunchuay, T. and Ketkaew, R. and Chotmongkolsap, P. and Chutimasakul, T. and Kanarat, J. and Tantirungrotechai, Y. and Tantirungrotechai, J.}, year = {2017}, pages = {6069--6079}, }